
Ein Yale-led collaborative study fördert das wissenschaftliche Verständnis, wie der Lungenerkrankung idiopathische pulmonale Fibrose (IPF) fortschreitet, bietet einen Fahrplan für Forscher entdecken neue Therapie-targets für die Krankheit.
Die Studie, geführt von Naftali Kaminski, M. D., der Boehringer-Ingelheim-stiftungsprofessor für Innere Medizin und Leiter der Abteilung der Lungen -, Kritischen Sorgfalt und der Schlafmedizin an der Yale School of Medicine und John E. McDonough, Lehrer und Forscher an der medical school, wird in JCI Einblick.
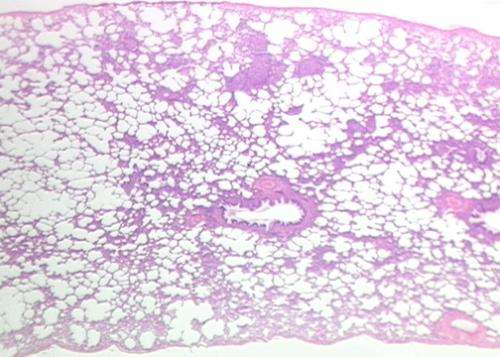
In der Studie untersuchten die Forscher unterschiedlich betroffenen Regionen in der Lunge, erhalten Sie von Personen mit IPF und gefunden, dass das, was aussieht wie normale Lunge ist bereits in Veränderungen in bestimmten Genen. Dann verfolgten Sie, wie diese Gene verändert, steigt oder sinkt, wie die Krankheit fortschreitet.
Ein einzigartiges Merkmal des Papiers, sagte Kaminski, ist, dass es die erste Computer-Modell des Fortschreitens der Krankheit in IPF-Lungen-und wird begleitet von einer interaktiven website mit der Erkundung dieses Modell. Kaminski glaubt, dass die breiten Zugang zu den Daten beschleunigen und die Forschung in neue Therapien bei IPF.
Obwohl Kaminski stellt fest, dass die Wissenschaftler an der Yale und anderswo gemacht haben „erhebliche wissenschaftliche Fortschritte“ bei IPF in den letzten Jahren, gibt es wenige Behandlungsmöglichkeiten. IPF ist eine chronische Erkrankung, bei der die Lunge zunehmend, vernarbt und nicht in der Lage zu funktionieren; es wirkt sich auf rund 200.000 Menschen in den USA, mit über 30.000 neue Fälle pro Jahr. Fünfzig Prozent der Patienten mit IPF sterben in drei bis fünf Jahren nach der Diagnose, und die Ursache der IPF unbekannt ist. Die zwei FDA-zugelassenen Medikamente zur Behandlung von IPF verlangsamen den Fortschritt der Krankheit, aber nicht umkehren. „Die Drogen kann nicht angenehm sein, aber Sie arbeiten“, sagte Kaminski, hinzufügen, dass, am wichtigsten ist, „Es gibt Hoffnung am Horizont.“
Arzneimittelstudien für die IPF sind im Gange, und dieser neuesten Forschung, sagte er, sollten Möglichkeiten für Forscher, um neue Angriffspunkte für Medikamente. „Meine Gruppe hat gefühlt seit Jahren, dass Interventionen zu entwickeln, für die IPF, die effektiver sind, müssen wir verstehen, wie die Krankheit fortschreitet in der menschlichen Lunge,“ Kaminski sagte.
Tiermodelle für IPF Arbeit, um zu zeigen, wie die pulmonale Fibrose in den Lungen, aber nicht das, was regelt änderungen auf genetischer Ebene zu fahren IPF-progression in den Menschen. Die Ermittler verwendet ein einzigartiges system, das Ihnen erlaubt zu quantifizieren, die Menge der Fibrose in der unterschiedlich betroffenen Regionen in der Lunge und dann zur Messung der expression aller Gene im menschlichen Genom, in genau der gleichen region, die durch RNA-Sequenzierung. Sie Maß auch microRNAs, kleine nicht kodierende RNAs bekannt, regulieren die expression von Genen. Sie angewendet erweiterte systembiologischen Methoden zu identifizieren, die Spuren der gen-expression im Zusammenhang mit der progression der IPF in der Lunge und die Moleküle regulieren. Mit diesem Ansatz machten Sie drei wichtige Erkenntnisse. Zuerst entdeckten Sie, dass das, was aussah wie normales Gewebe in die kranke Lunge war in der Tat abartig. Die zweite, Sie identifizierten das gen Ausdruck änderungen, die bestimmte waren zu einem Gewebe verbunden mit früh -, progressive-und end-Stufe Fibrose. Dritte, identifizierten Sie unterschiedliche molekulare Regulatoren für die einzelnen Stufen.
„Dies ist der erste Beweis, dass die verschiedenen Stadien der Erkrankung erfordern unterschiedliche Interventionen,“ sagte Kaminski.